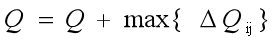哈尔滨工业大学
生物网络分析实践报告
学生姓名:荆树吉
学号: 25S103248
学院: 计算机科学与技术学院
**指导教师:**李杰
2026年 5月 12日
实验一: 基于蛋白互作网络的关键模块发现算法实现及功能注释 实验目的: 通过对整合的蛋白互作网络分析, 深刻理解识别生物网络关键模块的算法并对模块功能进行基因本体注释.
实验内容: 整合已有的蛋白互作网络资源和疾病相关基因,实现网络模块识别方法并识别重要的蛋白网络模块.
实验要求: 实现网络模块识别方法并能利用统计方法检验所识别的模块的统计显著性.
实验步骤: 1、从多个网站下载小的蛋白互作网络
2、将多个小的蛋白网络整合成一个大的蛋白互作网络
3、实现网络模块识别算法, 并利用这些算法从蛋白网络上识别出重要的蛋白网络模块
4、对关键模块进行生物功能注释
5、对关键模块进行显著性统计分析
6、编写网络可视化程序,将发现的重要功能模块用不同的颜色从网络上标示出来。
7、比较不同算法的网络模块分析结果,并分析差异的原因
实验算法: 1. Label propagation(标签传播算法) 算法:
每个节点赋予一个标签标志着其所在社区,每次迭代,每个节点标签根据其大多数邻近节点的标签而修改,收敛后具有相同标签的节点属于同一个社区。
算法步骤:
① 给每一个节点随机生成一个标签
② 随机生成一个所有节点的顺序,按照该顺序将每一个节点的标签修改为其大多数邻居节点的标签。
③ 重复②,直到每个节点的标签都不再变化,具有相同标签的节点组成了一个社区。
2. Newman Fast Algorithm(Newman快速算法) 算法:
对于具有N个节点的复杂网络,算法的执行包括以下步骤 :
① 初始化:初始化网络为N个社团,即每一个节点都是一个社团。
② 依次合并有边相连的社团,并计算合并后的模块度增量
③ 重复执行步骤②,不断合并社团,直到整个网络都合并为一个社团。
Newman快速算法属于凝聚算法的一种
3. DNE+层次聚类 本方法采用https://www.science.org/doi/10.1126/sciadv.adq4324,其中所介绍的点的扩展的神经网络方法生成每一个蛋白质的扩展向量,之后利用这些扩展向量使用层次聚类算法对于蛋白质网络进行聚类,词嵌入的流程大概为下图所示:
图 1 DNE的词汇嵌入生成流程
流程为:
1.输入是一个图网络(节点 + 边)。
每个节点先用图拉普拉斯特征初始化:
计算邻接矩阵的拉普拉斯特征(Laplacian Eigenvectors, LE)
这部分体现节点在全局结构中的位置
2.结构化采样:
选一个“锚点节点”(anchor)
正样本:从该锚点的邻居中采样,表示局部结构相似
负样本:从远离锚点的节点中采样,通常基于节点度分布来挑选,表示非局部或不相似节点
3.DNE 预训练:
使用编码器把节点特征映射到低维嵌入空间
编码器输入包括:
LE 结构特征
(可选)节点的额外特征
训练目标是对比学习:
①让锚点与正样本在嵌入空间靠近
②让锚点与负样本在嵌入空间远离
在进行完词嵌入的生成之后再将结果输入对应的分层聚类方法,之后生成聚类结果。
实验环境 :
R-4.5.2 library(igraph) library(dplyr) library(readr),Python3.10.18
Python包
import os
import sys
from pathlib import Path
import numpy as np
import pandas as pd
import networkx as nx
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
from sklearn.cluster import AgglomerativeClustering
from sklearn.metrics import silhouette_score
实验数据: 511145.protein.physical.links.v12.0.min900.txt
链接:
https://stringdb-downloads.org/download/stream/protein.links.full.v12.0/511145.protein.links.full.v12.0.min900.txt.gz
本研究采用 STRING v12.0 数据库中 大肠杆菌(Taxonomy ID: 511145) 的全量蛋白互作数据,筛选综合置信度≥0.900的高可信度互作对,构建无向加权蛋白互作(PPI)网络。数据以边列表格式存储,包含蛋白编号与互作置信度信息,经去重、去自环预处理后用于后续社区挖掘与拓扑分析,总结点数:1293,总边数:4402。
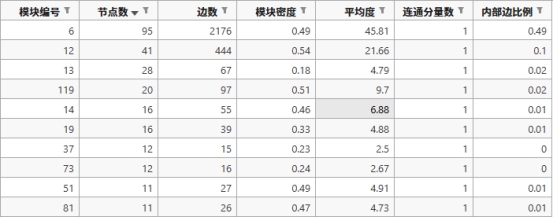
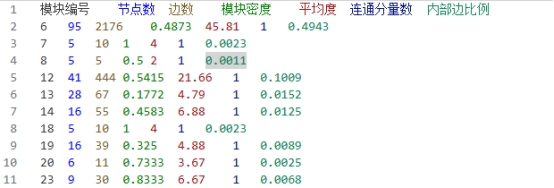
实验结果及分析: 1. Label propagation(标签传播算法) ① 部分示例 下面是标签传播算法分析出来的比较重要的一个模块
模块编号 节点数 边数 模块密度 平均度 连通分量数 内部边比例
6
95
2176
0.4873
45.81
1
0.4943
图 2 标签传播法的集群截图
上面是标签传播发所产生的模块6的对应的各项数据和一些蛋白质。
② 关键模块注释 经过标签传播算法分析之后,整体的模块数为325块,其中有76块模块的模块度大于整体模块度且对应的节点数大于等于5,这些重要模块的数量比较大,且其分块数据带有511145.的前缀,不可以直接进行GO的富集注释,因此经python代码进行处理后,还需要进一步的筛选,我选择了在重要模块中节点数量前十的模块作为关键模块进行注释。
图 3 前十的LPA关键模块的特征
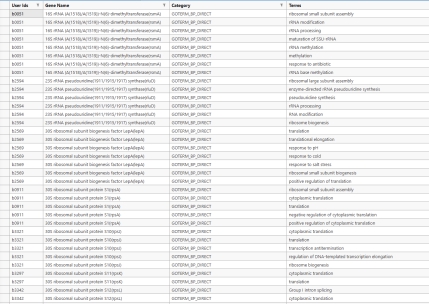
以模块六为例,下图是筛选并截取过后的模块的对应的基因(其实也是通用的蛋白质)编号。
图 4 核心模块6的聚类结果(截取过后)
将结果输入到David GO在线注释网站上,生成注释结果(主要关注模块的BP也就是对应的生物过程)。操作过程示意如下。
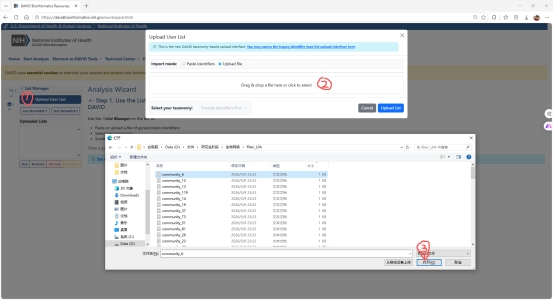
图 5 David GO文件上传
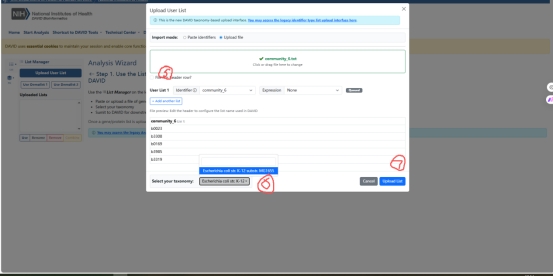
图 6 David GO参数处理
文件上传到David GO之后要选取对应的生物体,我的生物体是K12大肠杆菌,之后就可以进行分析了。
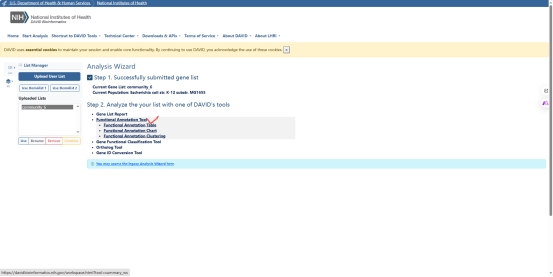
图 7 David GO分析
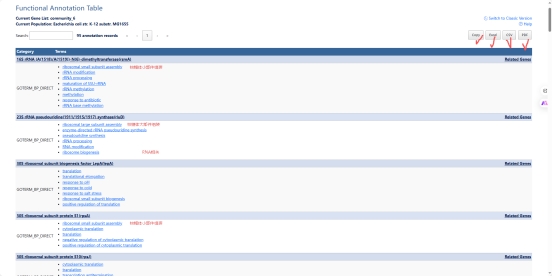
图 8 GO 富集注释
David GO选择富集注释,并且选择对应的最直接,最第一个级别的注释结果,这样的生物过程注释最紧密,最严谨。
图 9 GO 富集注释直接(最近概念)注释
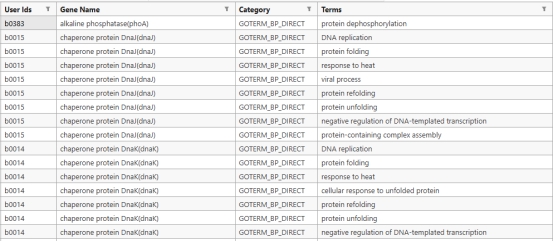
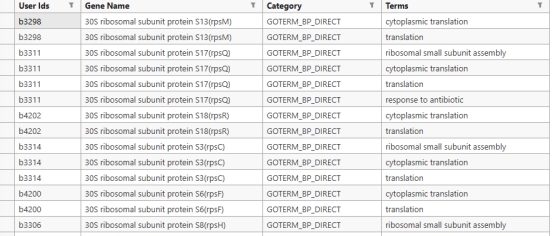
GO的富集注释结果可以导出也可以直接在网站上进行观察,比如这份LPA的第六个模块的蛋白质(基因)注释结果,这个模块注释是大肠杆菌核糖体 及蛋白翻译 相关基因的GO生物学功能注释,记录了每个基因编码的蛋白直接参与核糖体组装 、rRNA修饰 、蛋白翻译 全程及胁迫应答、蛋白转运 等多项生理功能,整体构成细菌完整蛋白质合成系统的关键基因集合(其余结果在文件中无法全部展示)。
图 10 关键模块6注释(LPA)导出
上图是导出之后的关键模块的注释表格。
图 11 DAG(LPA module 6)
上图是根据第六个模块的注释生成的带有基因标注的有向无环图,其中的根节点就是对应的最小概念功能。
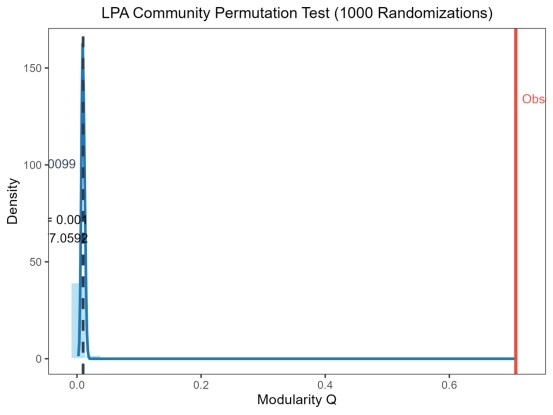
③ 统计分析 总结点数:1293,总边数:4402,总模块数325,真实网络的LPA模块度Q=0.7071,置换检验1000次,全局显著值P=0.001,随机网络平均Q=0.0099,随机网络Q标准差=0.0025,Z-score=277.0592,结果判定LPA模块结构显著。下面是对应的部分的显著模块的统计学信息。
图 12 LPA部分显著模块的统计学特征
下面是千次检验对应的直方图与实际LPA聚类的对比图,可以看到效果很良好。
图 13 LPA千次检验直方图
④ 可视化重要模块
图 14 蛋白质互作LPA示意图
在这一部分,我筛选出了包含的节点大于5,并且模块度大于整体模块度的模块进行可视化。
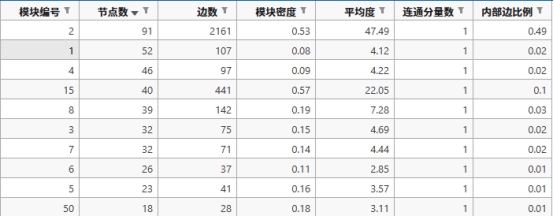
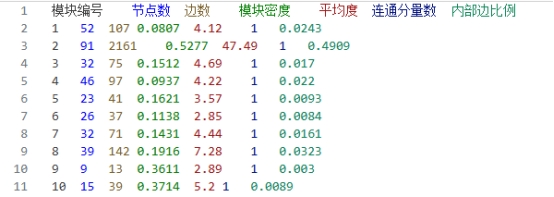
2. Newman Fast Algorithm(Newman快速算法) ① 部分示例 下面是Newman快速算法分析出来的比较重要的一个模块
模块编号 节点数 边数 模块密度 平均度 连通分量数 内部边比例
2
91
2161
0.5277
47.49
1
0.4909
图 15 Newman快速算法的集群截图
② 关键模块注释 经过Newman快速算法分析之后,整体的模块数为293块,其中有59块模块的模块度大于整体模块度且对应的节点数大于等于5,同样我选择了在重要模块中节点数量前十的模块作为关键模块进行GO富集注释。
图 16 前十的NFA关键模块的特征
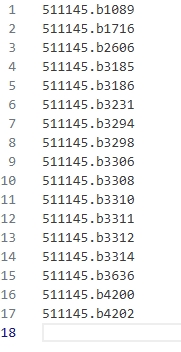
以模块一为例(对应的最大的模块注释与LPA相似,不再使用这个模块),下图是筛选并截取过后的模块的对应的基因(其实也是通用的蛋白质)编号。
图 17 核心模块1的聚类结果(截取过后)
将结果输入到David GO在线注释网站上,生成注释结果(关注模块的BP也就是对应的生物过程)。操作过程示意与LPA的注释相同。
图 18 关键模块1注释(NFA)导出
上面是NFA导出的关键模块1的注释结果,这份文档是大肠杆菌膜转运、外膜组装、蛋白折叠分泌及离子营养转运 相关基因的GO功能注释。
图 19 DAG(NFA module 1)
上图是根据第1个模块的注释生成的带有基因标注的有向无环图,其中的根节点就是对应的最小概念功能。
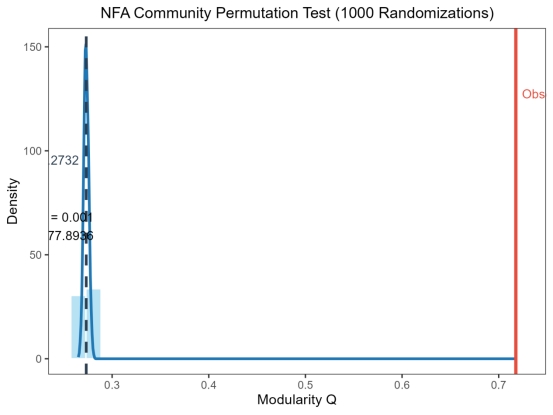
③ 统计分析 总结点数:1293,总边数:4402,总模块数293,真实网络的NFA模块度Q=0.7177,置换检验1000次,全局显著值P=0.001,随机网络平均Q=0.2732,随机网络Q标准差=0.0025,Z-score=177.8936,结果判定NFA模块结构显著。下面是对应的部分的显著模块的统计学信息。
图 20 NFA部分显著模块的统计学特征
下面是NFA千次检验的置信度测试图表,可以看到,效果依旧很好。
图 21 NFA千次检验直方图
④ 可视化重要模块
图 22 蛋白质互作NFA示意图
在这一部分,和上一个方法的控制方式相同。
3.DNE+层次聚类 ①部分示例 下面是DNE+层次聚类分析出来的比较重要的一个模块
模块编号 节点数 边数 模块密度 平均度 连通分量数 内部边比例
4
17
136
1
16
1
0.0413
图 23 DNE+层次聚类的集群截图
②关键模块注释 经过DNE+层次聚类算法分析之后,整体的模块数为400块,其中有33块模块的模块度大于整体模块度且对应的节点数大于等于5,同样我选择了在重要模块中节点数量前十的模块作为关键模块进行GO富集注释。
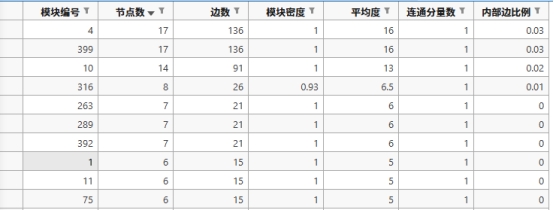
图 24 前十的DNE关键模块的特征
以模块4为例下图是筛选并截取过后的模块的对应的基因(其实也是通用的蛋白质)编号。
图 25 核心模块0的聚类结果(截取过后)
将结果输入到David GO在线注释网站上,生成注释结果(关注模块的BP也就是对应的生物过程)。操作过程示意与LPA的注释相同。
图 26 关键模块4注释(DNE)导出
上面是DNE导出的关键模块4的注释结果,该基因集群均编码核糖体大小亚基结构蛋白,主要参与核糖体亚基组装、胞质内蛋白质翻译核心过程,同时兼具翻译正负调控、mRNA 稳定性调节功能,还可响应抗生素、氧化应激与辐射胁迫,也参与细胞生长及核糖体生物合成调控。
上图是根据第4个模块的注释与LPA最大的模块相似,略过。
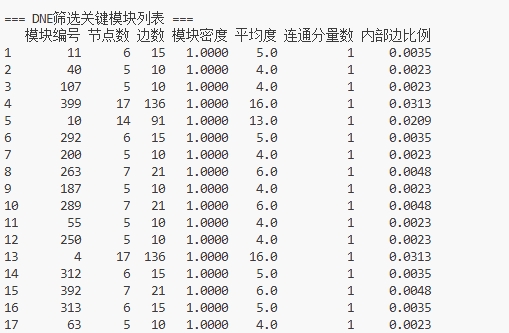
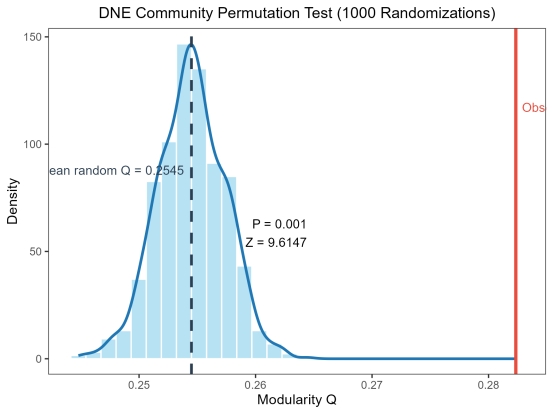
③统计分析 总结点数:1293,总边数:4347,总模块数400,真实网络的NFA模块度Q=0.2823,置换检验1000次,全局显著值P=0.001,随机网络平均Q=0.2545,随机网络Q标准差=0.0029,Z-score=9.6147,结果判定DNE模块结构显著。下面是对应的部分的显著模块的统计学信息。
图 27 DNE部分显著模块的统计学特征
下面是DNE千次检验的置信度测试图表,可以看到,效果比较合适。
图 28 DNE千次检验直方图
④可视化重要模块
图 29 蛋白质互作GNE示意图
在这一部分,和上一个方法的控制方式相同。
总结 DNE为向量层次聚类,LPA是标签传播算法,NFA为Newman快速社区算法,三者聚类结果差异显著。
DNE依据向量相似度逐层合并聚类,聚类数量可人为截断控制,簇呈现嵌套层级结构,易形成规模差距较大的簇,低相似样本易单独成小簇。
LPA依靠邻域标签迭代扩散划分社区,聚类结果随机性较强,易产生细碎社区与孤立节点,边界交叉模糊,边缘节点易被邻近群落同化。NFA以最大化模块度为贪心目标划分社区,最终聚类数目固定,社区内部连接紧密、外部关联稀疏,簇划分规整均衡,节点归属稳定。
结果差异源于算法原理不同:DNE以空间向量距离为聚类依据;LPA依托拓扑关系随机标签更新,无优化目标约束;NFA全局优化模块度,追求网络社区结构最优。三类算法分别适配多维层级数据、无参网络划分、最优结构社区挖掘场景。
附件-实验一程序和数据 DNE+层次聚类: module_detection_dne.ipynb 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 这个 notebook 演示如何对 `data/a_thaliana/511145. protein.physical.links.v12.0 .min900.txt` 进行模块检测, 并输出: - 每个模块包含的蛋白质列表文件 - 按聚类编号排序的蛋白质-聚类结果文件 - 网络可视化图像(模块颜色 + 节点度大小) import os import sys from pathlib import Path import numpy as npimport pandas as pdimport networkx as nximport matplotlib.pyplot as pltimport matplotlib.colors as mcolorsfrom sklearn.cluster import AgglomerativeClusteringfrom sklearn.metrics import silhouette_scoreimport igraph as igsys.path.append('code' ) from models.dne import DNEoutput_dir = Path("D:/文件/研究生阶段/生物网络/实验1蛋白质模块发现/data/DNE_cengci_Community_Proteins" ) os.makedirs(output_dir, exist_ok=True ) ppi_path = Path('D:/文件/研究生阶段/生物网络/实验1蛋白质模块发现/data/511145.protein.physical.links.v12.0.min900.txt' ) print ('output_dir =' , output_dir)print ('ppi_path =' , ppi_path.resolve())try : edges = pd.read_csv(ppi_path, sep=r'\s+' , header=None , comment='#' , usecols=[0 , 1 ], names=['source' , 'target' ], engine='python' ) except Exception as ex: raise RuntimeError(f'读取文件失败,请检查文件格式: {ex} ' ) edges = edges.dropna(subset=['source' , 'target' ]).astype(str ) edges = edges[edges['source' ] != edges['target' ]] edges = edges.drop_duplicates() G = nx.Graph() G.add_edges_from(edges[['source' , 'target' ]].itertuples(index=False , name=None )) G.remove_nodes_from(list (nx.isolates(G))) print (f'节点数: {G.number_of_nodes()} ' )print (f'边数: {G.number_of_edges()} ' )print (f'是否连通: {nx.is_connected(G)} ' )summary = pd.DataFrame({ 'stat' : ['num_nodes' , 'num_edges' , 'connected' ], 'value' : [G.number_of_nodes(), G.number_of_edges(), nx.is_connected(G)] }) summary.to_csv(output_dir / 'graph_summary.csv' , index=False ) model = DNE(G, hidden_dim=128 , num_pos_features=256 ) train_params = { 'batch_size' : 5120 , 'epochs' : 10 , 'walk_number' : 30 , 'walk_length' : 10 , 'p' : 1.0 , 'q' : 1.0 , } print ('开始训练 DNE,可能需要几分钟...' )model.train(**train_params) print ('DNE 训练完成' )使用 DNE 学到的嵌入进行模块检测。 embeddings = model.get_embeddings() node_list = sorted (embeddings.keys()) X = np.vstack([embeddings[node] for node in node_list]) best_n_clusters = 2 best_score = -1 max_clusters = min (400 , len (node_list) - 1 ) print (f'开始自动调优聚类数,从 2 到 {max_clusters} ...' )for n_clusters in range (2 , max_clusters + 1 ): cluster_model = AgglomerativeClustering(n_clusters=n_clusters) cluster_labels = cluster_model.fit_predict(X) score = silhouette_score(X, cluster_labels) if score > best_score: best_score = score best_n_clusters = n_clusters if n_clusters % 50 == 0 : print (f'已测试到 {n_clusters} 个簇,最佳目前: {best_n_clusters} (轮廓系数: {best_score:.4 f} )' ) print (f'最佳聚类数: {best_n_clusters} ,轮廓系数: {best_score:.4 f} ' )cluster_model = AgglomerativeClustering(n_clusters=best_n_clusters) cluster_labels = cluster_model.fit_predict(X) cluster_df = pd.DataFrame({ 'protein_name' : node_list, 'cluster_id' : cluster_labels }) cluster_df = cluster_df.sort_values(['cluster_id' , 'protein_name' ]).reset_index(drop=True ) cluster_df.to_csv(output_dir / 'a_Cluster_Info.txt' , index=False ,header=False , sep='\t' ) print ('已保存 a_Cluster_Info.txt' )for cluster_id, group in cluster_df.groupby('cluster_id' ): module_path = output_dir / f'community_{cluster_id} .txt' group['protein_name' ].to_csv(module_path, index=False , header=False ) print ('已保存每个模块的单独文件' )net_density = nx.density(G) module_stats = [] for cluster_id in cluster_df['cluster_id' ].unique(): module_nodes = cluster_df[cluster_df['cluster_id' ] == cluster_id]['protein_name' ].tolist() if len (module_nodes) >= 5 : subgraph = G.subgraph(module_nodes) module_density = nx.density(subgraph) if module_density > net_density: module_stats.append({ 'cluster_id' : cluster_id, 'nodes' : module_nodes, 'density' : module_density, 'size' : len (module_nodes) }) print (f'整体网络密度: {net_density:.4 f} ' )print (f'筛选出 {len (module_stats)} 个关键模块(节点数>=5且密度>整体网络密度)' )
vis.R 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 if ( ! require( igraph) ) install.packages( "igraph" ) library( igraph) df <- read.table( "data/511145.protein.physical.links.v12.0.min900.txt" , header = F , sep = " " , stringsAsFactors = F ) df_edge <- df[ , 1 : 2 ] df_edge <- na.omit( df_edge) df_edge <- unique( df_edge) g <- graph_from_edgelist( as.matrix( df_edge) , directed = FALSE ) gene_cluster_df <- read.table( "data/DNE_cengci_Community_Proteins/a_Cluster_Info.txt" , sep = "\t" , header = FALSE , stringsAsFactors = FALSE ) colnames( gene_cluster_df) <- c ( "Gene" , "Cluster" ) all_genes <- names ( V( g) ) V( g) $ community <- gene_cluster_df$ Cluster[ match( all_genes, gene_cluster_df$ Gene) ] global_dens <- edge_density( g) member_all <- V( g) $ community keep_comm <- c ( ) for ( comm_id in unique( member_all) ) { nodes_idx <- which( member_all == comm_id) if ( length ( nodes_idx) > 5 ) { subg <- induced_subgraph( g, nodes_idx) comm_dens <- edge_density( subg) if ( comm_dens > global_dens) { keep_comm <- c ( keep_comm, comm_id) } } } plot_nodes <- which( member_all %in% keep_comm) g_plot <- induced_subgraph( g, plot_nodes) V( g_plot) $ community <- member_all[ plot_nodes] deg <- degree( g_plot) V( g_plot) $ size <- sqrt ( deg) * 1.2 n_comm <- length ( unique( V( g_plot) $ community) ) pal <- rainbow( n_comm) V( g_plot) $ color <- pal[ as.factor( V( g_plot) $ community) ] E( g_plot) $ color <- "gray30" E( g_plot) $ width <- 1 node_names <- names ( V( g_plot) ) V( g_plot) $ short_label <- substr( node_names, nchar( node_names) - 3 , nchar( node_names) ) lay <- layout_with_fr( g_plot) scale_factor <- 3.5 lay <- lay * scale_factor plot( g_plot, main = "基因互作网络 DNE+层次聚类 社区聚类图" , layout = lay, vertex.label = V( g_plot) $ short_label, vertex.label.cex = 0.28 , vertex.label.dist = 1.1 , vertex.label.color = "black" , edge.color = E( g_plot) $ color, edge.width = E( g_plot) $ width, margin = c ( 0.4 , 0.4 , 0.4 , 0.4 ) ) png( "png/GeneNetwork_DNE_protein.png" , width = 4000 , height = 4000 , res = 300 ) plot( g_plot, main = "基因互作网络 DNE+层次聚类 社区聚类图" , layout = lay, vertex.label = V( g_plot) $ short_label, vertex.label.cex = 0.28 , vertex.label.dist = 1.1 , vertex.label.color = "black" , edge.color = E( g_plot) $ color, edge.width = E( g_plot) $ width) dev.off( ) cat( "✅ 已读取已有聚类元数据,按原筛选规则+原绘图样式完成重绘并保存图片\n" )
statistic_DNE_R 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 library( igraph) library( dplyr) library( readr) library( ggplot2) set.seed( 123 ) edge_file <- "data/511145.protein.physical.links.v12.0.min900.txt" cluster_file <- "data/DNE_cengci_Community_Proteins/a_Cluster_Info.txt" edges <- read_delim( edge_file, delim = " " , col_names = FALSE , show_col_types = FALSE ) %>% select( 1 , 2 ) colnames( edges) <- c ( "protein1" , "protein2" ) clusters <- read_delim( cluster_file, delim = "\t" , col_names = TRUE , show_col_types = FALSE ) colnames( clusters) <- c ( "protein" , "module" ) g <- graph_from_data_frame( edges, directed = FALSE ) g <- simplify( g, remove.multiple = TRUE , remove.loops = TRUE ) V( g) $ module <- clusters$ module[ match( V( g) $ name, clusters$ protein) ] g <- induced_subgraph( g, ! is.na ( V( g) $ module) ) cat( "=== 网络基础信息 ===\n" ) cat( "总节点数:" , vcount( g) , "\n" ) cat( "总边数:" , ecount( g) , "\n" ) nfa_module_num <- length ( unique( V( g) $ module) ) cat( "总模块数(DNE划分):" , nfa_module_num, "\n\n" ) cat( "=== DNE全局模块显著性分析 ===\n" ) real_module <- make_clusters( g, V( g) $ module) real_Q <- modularity( real_module) cat( "真实网络 DNE 模块化度 Q =" , round ( real_Q, 4 ) , "\n" ) n_perm <- 1000 random_Q <- numeric( n_perm) for ( i in 1 : n_perm) { g_random <- rewire( g, keeping_degseq( niter = vcount( g) * 5 ) ) dne_hc <- cluster_fast_greedy( g_random) memb_vec <- cut_at( dne_hc, no = nfa_module_num) rand_clu <- make_clusters( g_random, memb_vec) random_Q[ i] <- modularity( rand_clu) if ( i %% 200 == 0 ) cat( "已完成" , i, "次随机置换检验\n" ) } p_raw <- mean( random_Q >= real_Q) p_value <- ifelse( p_raw == 0 , 1 / ( n_perm + 1 ) , p_raw) cat( "全局显著性 P值 =" , ifelse( p_raw == 0 , paste0( round ( p_value, 4 ) , " (P < 0.001)" ) , round ( p_value, 4 ) ) , "\n" ) cat( "随机网络平均 Q =" , round ( mean( random_Q) , 4 ) , "\n" ) cat( "随机网络 Q 标准差 =" , round ( sd( random_Q) , 4 ) , "\n" ) cat( "Z-score =" , round ( ( real_Q - mean( random_Q) ) / sd( random_Q) , 4 ) , "\n" ) cat( "结果判定:" , ifelse( p_value < 0.05 , "✅ DNE模块结构显著(非随机)" , "❌ DNE模块结构无统计学显著性" ) , "\n\n" ) write.csv( data.frame( random_Q) , "data/DNE_cengci_Community_Proteins/random_Q.csv" , row.names = FALSE ) module_stats <- lapply( unique( V( g) $ module) , function ( m) { module_nodes <- V( g) $ name[ V( g) $ module == m] subg <- induced_subgraph( g, module_nodes) intra_edges <- ecount( subg) total_edges_all <- ecount( g) data.frame( 模块编号 = m, 节点数 = vcount( subg) , 边数 = ecount( subg) , 模块密度 = round ( edge_density( subg) , 4 ) , 平均度 = round ( mean( degree( subg) ) , 2 ) , 连通分量数 = components( subg) $ no, 内部边比例 = round ( intra_edges / total_edges_all, 4 ) ) } ) %>% bind_rows( ) %>% arrange( desc( 模块密度) ) net_density <- edge_density( g) key_modules <- module_stats %>% filter( 节点数 >= 5 , 模块密度 > net_density, 连通分量数 == 1 ) cat( "=== DNE筛选关键模块列表 ===\n" ) print( key_modules) write_delim( arrange( module_stats, 模块编号) , "data/DNE_cengci_Community_Proteins/aDNE_蛋白质模块全统计结果.txt" , delim = "\t" ) write_delim( arrange( key_modules, 模块编号) , "data/DNE_cengci_Community_Proteins/aDNE_关键蛋白质模块筛选结果.txt" , delim = "\t" ) cat( "\n✅ DNE方法专属分析完成!统计结果已保存至 DNE_cengci_Community_Proteins 目录" ) plot_df <- data.frame( Q_value = random_Q) p <- ggplot( plot_df, aes( x = Q_value) ) + geom_histogram( aes( y = ..density..) , bins = 30 , fill = "#87CEEB" , alpha = 0.6 , color = "white" ) + geom_density( color = "#1f77b4" , linewidth = 1 ) + geom_vline( xintercept = real_Q, color = "#e74c3c" , linewidth = 1.2 , linetype = "solid" ) + geom_vline( xintercept = mean( random_Q) , color = "#2c3e50" , linewidth = 1 , linetype = "dashed" ) + annotate( "text" , x = real_Q, y = max ( density( random_Q) $ y) * 0.8 , label = paste0( "Observed Q = " , round ( real_Q, 4 ) ) , color = "#e74c3c" , hjust = - 0.05 , size = 3.5 ) + annotate( "text" , x = mean( random_Q) , y = max ( density( random_Q) $ y) * 0.6 , label = paste0( "Mean random Q = " , round ( mean( random_Q) , 4 ) ) , color = "#2c3e50" , hjust = 1.05 , size = 3.5 ) + annotate( "text" , x = max ( random_Q) , y = max ( density( random_Q) $ y) * 0.4 , label = paste0( "P = " , round ( p_value, 4 ) , "\nZ = " , round ( ( real_Q - mean( random_Q) ) / sd( random_Q) , 4 ) ) , size = 3.5 , hjust = 1 ) + labs( x = "Modularity Q" , y = "Density" , title = "DNE Community Permutation Test (1000 Randomizations)" ) + theme_bw( ) + theme( plot.title = element_text( hjust = 0.5 , size = 12 ) , axis.title = element_text( size = 11 ) , panel.grid = element_blank( ) ) print( p) ggsave( "png/DNE_Permutation_Test_Q.png" , plot = p, width = 6 , height = 4.5 , dpi = 300 )
LPA 标签传播检验 标签传播法.R 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 if ( ! require( igraph) ) install.packages( "igraph" ) library( igraph) df <- read.table( "data/511145.protein.physical.links.v12.0.min900.txt" , header = F , sep = " " , stringsAsFactors = F ) df_edge <- df[ , 1 : 2 ] df_edge <- na.omit( df_edge) df_edge <- unique( df_edge) g <- graph_from_edgelist( as.matrix( df_edge) , directed = FALSE ) set.seed( 123 ) lpa_comm <- cluster_label_prop( g) cat( "=====================================\n" ) cat( "📊 网络基础信息统计:\n" ) cat( "总节点数量:" , vcount( g) , "\n" ) cat( "总边数量:" , ecount( g) , "\n" ) cat( "=====================================\n" ) lpa_mod <- modularity( lpa_comm) cat( "\n=====================================\n" ) cat( "LPA 最优聚类对应的模块度 Q = " , round ( lpa_mod, 4 ) , "\n" ) if ( lpa_mod > 0.3 ) { cat( "👉 模块度 > 0.3,网络社区结构显著\n" ) } else { cat( "👉 模块度 ≤ 0.3,网络社区结构较弱\n" ) } cat( "=====================================\n" ) cat( "各社区成员数量:\n" ) print( sizes( lpa_comm) ) V( g) $ community <- membership( lpa_comm) gene_name <- names ( V( g) ) cluster_id <- membership( lpa_comm) gene_cluster_df <- data.frame( Gene = gene_name, Cluster = cluster_id, row.names = NULL ) if ( ! dir.exists( "data/LPA_Community_Proteins" ) ) { dir.create( "data/LPA_Community_Proteins" ) } write.table( gene_cluster_df, file = "data/LPA_Community_Proteins/a_Cluster_Info.txt" , sep = "\t" , quote = FALSE , row.names = FALSE , col.names = FALSE ) cat( "\n✅ 已输出:a_Cluster_Info.txt (第1列基因名,第2列集群编号)\n" ) comm_list <- split( names ( V( g) ) , membership( lpa_comm) ) for ( i in seq_along ( comm_list) ) { gene_vec <- comm_list[[ i] ] writeLines( gene_vec, con = paste0( "data/LPA_Community_Proteins/community_" , i, ".txt" ) ) } cat( "\n✅ 已将每个聚类群体节点名分别保存到 LPA_Community_Proteins 文件夹\n" ) global_dens <- edge_density( g) member_all <- membership( lpa_comm) keep_comm <- c ( ) for ( comm_id in unique( member_all) ) { nodes_idx <- which( member_all == comm_id) if ( length ( nodes_idx) > 5 ) { subg <- induced_subgraph( g, nodes_idx) comm_dens <- edge_density( subg) if ( comm_dens > global_dens) { keep_comm <- c ( keep_comm, comm_id) } } } plot_nodes <- which( member_all %in% keep_comm) g_plot <- induced_subgraph( g, plot_nodes) V( g_plot) $ community <- member_all[ plot_nodes] deg <- degree( g_plot) V( g_plot) $ size <- sqrt ( deg) * 1.2 n_comm <- length ( unique( V( g_plot) $ community) ) pal <- rainbow( n_comm) V( g_plot) $ color <- pal[ as.factor( V( g_plot) $ community) ] E( g_plot) $ color <- "gray30" E( g_plot) $ width <- 1 node_names <- names ( V( g_plot) ) V( g_plot) $ short_label <- substr( node_names, nchar( node_names) - 3 , nchar( node_names) ) lay <- layout_with_fr( g_plot) scale_factor <- 3.5 lay <- lay * scale_factor plot( g_plot, main = "基因互作网络 LPA 社区聚类图" , layout = lay, vertex.label = V( g_plot) $ short_label, vertex.label.cex = 0.28 , vertex.label.dist = 1.1 , vertex.label.color = "black" , edge.color = E( g_plot) $ color, edge.width = E( g_plot) $ width, margin = c ( 0.4 , 0.4 , 0.4 , 0.4 ) ) png( "png/GeneNetwork_LPA_protein.png" , width = 4000 , height = 4000 , res = 300 ) plot( g_plot, main = "基因互作网络 LPA 社区聚类图" , layout = lay, vertex.label = V( g_plot) $ short_label, vertex.label.cex = 0.28 , vertex.label.dist = 1.1 , vertex.label.color = "black" , edge.color = E( g_plot) $ color, edge.width = E( g_plot) $ width) dev.off( ) cat( "\n✅ 可视化仅展示:节点数>5 且 模块密度优于全局的模块,图片已保存\n" )
statistic_LPA_.R 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 library( igraph) library( dplyr) library( readr) set.seed( 123 ) edge_file <- "data/511145.protein.physical.links.v12.0.min900.txt" cluster_file <- "data/LPA_Community_Proteins/a_Cluster_Info.txt" edges <- read_delim( edge_file, delim = " " , col_names = FALSE , show_col_types = FALSE ) %>% select( 1 , 2 ) colnames( edges) <- c ( "protein1" , "protein2" ) clusters <- read_delim( cluster_file, delim = "\t" , col_names = FALSE , show_col_types = FALSE ) colnames( clusters) <- c ( "protein" , "module" ) g <- graph_from_data_frame( edges, directed = FALSE ) g <- simplify( g, remove.multiple = TRUE , remove.loops = TRUE ) V( g) $ module <- clusters$ module[ match( V( g) $ name, clusters$ protein) ] g <- induced_subgraph( g, ! is.na ( V( g) $ module) ) cat( "=== 网络基础信息 ===\n" ) cat( "总节点数:" , vcount( g) , "\n" ) cat( "总边数:" , ecount( g) , "\n" ) lpa_module_num <- length ( unique( V( g) $ module) ) cat( "总模块数(LPA划分):" , lpa_module_num, "\n\n" ) cat( "=== LPA全局模块显著性分析 ===\n" ) real_module <- make_clusters( g, V( g) $ module) real_Q <- modularity( real_module) cat( "真实网络 LPA 模块化度 Q =" , round ( real_Q, 4 ) , "\n" ) n_perm <- 1000 random_Q <- numeric( n_perm) for ( i in 1 : n_perm) { g_random <- rewire( g, keeping_degseq( niter = vcount( g) * 5 ) ) lpa_clu <- cluster_label_prop( g_random) memb_vec <- membership( lpa_clu) rand_clu <- make_clusters( g_random, memb_vec) random_Q[ i] <- modularity( rand_clu) if ( i %% 200 == 0 ) cat( "已完成" , i, "次随机置换检验\n" ) } p_raw <- mean( random_Q >= real_Q) p_value <- ifelse( p_raw == 0 , 1 / ( n_perm + 1 ) , p_raw) cat( "全局显著性 P值 =" , round ( p_value, 4 ) , "\n" ) cat( "随机网络平均 Q =" , round ( mean( random_Q) , 4 ) , "\n" ) cat( "随机网络 Q 标准差 =" , round ( sd( random_Q) , 4 ) , "\n" ) cat( "Z-score =" , round ( ( real_Q - mean( random_Q) ) / sd( random_Q) , 4 ) , "\n" ) cat( "结果判定:" , ifelse( p_value < 0.05 , "✅ LPA模块结构显著(非随机)" , "❌ LPA模块结构无统计学显著性" ) , "\n\n" ) write.csv( data.frame( random_Q) , "data/LPA_Community_Proteins/random_Q.csv" , row.names = FALSE ) module_stats <- lapply( unique( V( g) $ module) , function ( m) { module_nodes <- V( g) $ name[ V( g) $ module == m] subg <- induced_subgraph( g, module_nodes) intra_edges <- ecount( subg) total_edges_all <- ecount( g) data.frame( 模块编号 = m, 节点数 = vcount( subg) , 边数 = ecount( subg) , 模块密度 = round ( edge_density( subg) , 4 ) , 平均度 = round ( mean( degree( subg) ) , 2 ) , 连通分量数 = components( subg) $ no, 内部边比例 = round ( intra_edges / total_edges_all, 4 ) ) } ) %>% bind_rows( ) %>% arrange( desc( 模块密度) ) net_density <- edge_density( g) key_modules <- module_stats %>% filter( 节点数 >= 5 , 模块密度 > net_density, 连通分量数 == 1 ) cat( "=== LPA筛选关键模块列表 ===\n" ) print( key_modules) write_delim( arrange( module_stats, 模块编号) , "data/LPA_Community_Proteins/aLPA_蛋白质模块全统计结果.txt" , delim = "\t" ) write_delim( arrange( key_modules, 模块编号) , "data/LPA_Community_Proteins/aLPA_关键蛋白质模块筛选结果.txt" , delim = "\t" ) cat( "\n✅ LPA方法专属分析完成!统计结果已保存至 LPA_Community_Proteins 目录" ) plot_df <- data.frame( Q_value = random_Q) p <- ggplot( plot_df, aes( x = Q_value) ) + geom_histogram( aes( y = ..density..) , bins = 30 , fill = "#87CEEB" , alpha = 0.6 , color = "white" ) + geom_density( color = "#1f77b4" , linewidth = 1 ) + geom_vline( xintercept = real_Q, color = "#e74c3c" , linewidth = 1.2 , linetype = "solid" ) + geom_vline( xintercept = mean( random_Q) , color = "#2c3e50" , linewidth = 1 , linetype = "dashed" ) + annotate( "text" , x = real_Q, y = max ( density( random_Q) $ y) * 0.8 , label = paste0( "Observed Q = " , round ( real_Q, 4 ) ) , color = "#e74c3c" , hjust = - 0.05 , size = 3.5 ) + annotate( "text" , x = mean( random_Q) , y = max ( density( random_Q) $ y) * 0.6 , label = paste0( "Mean random Q = " , round ( mean( random_Q) , 4 ) ) , color = "#2c3e50" , hjust = 1.05 , size = 3.5 ) + annotate( "text" , x = max ( random_Q) , y = max ( density( random_Q) $ y) * 0.4 , label = paste0( "P = " , round ( p_value, 4 ) , "\nZ = " , round ( ( real_Q - mean( random_Q) ) / sd( random_Q) , 4 ) ) , size = 3.5 , hjust = 1 ) + labs( x = "Modularity Q" , y = "Density" , title = "LPA Community Permutation Test (1000 Randomizations)" ) + theme_bw( ) + theme( plot.title = element_text( hjust = 0.5 , size = 12 ) , axis.title = element_text( size = 11 ) , panel.grid = element_blank( ) ) print( p) ggsave( "png/LPA_Permutation_Test_Q.png" , plot = p, width = 6 , height = 4.5 , dpi = 300 )
NFA Newman Fas算法 NFA.R 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 if ( ! require( igraph) ) install.packages( "igraph" ) library( igraph) library( dplyr) df <- read.table( "data/511145.protein.physical.links.v12.0.min900.txt" , header = F , sep = " " , stringsAsFactors = F ) df_edge <- df[ , 1 : 2 ] df_edge <- na.omit( df_edge) df_edge <- unique( df_edge) g <- graph_from_edgelist( as.matrix( df_edge) , directed = FALSE ) g <- simplify( g, remove.multiple = TRUE , remove.loops = TRUE ) set.seed( 123 ) nfa_comm <- cluster_fast_greedy( g) cat( "Newman Fast Algorithm (NFA) 层次聚类树状结构:\n" ) print( nfa_comm) nfa_best <- membership( nfa_comm) cat( "=====================================\n" ) cat( "📊 网络基础信息统计:\n" ) cat( "总节点数量:" , vcount( g) , "\n" ) cat( "总边数量:" , ecount( g) , "\n" ) cat( "=====================================\n" ) nfa_mod <- modularity( nfa_comm) cat( "\n=====================================\n" ) cat( "NFA 最优聚类对应的模块度 Q = " , round ( nfa_mod, 4 ) , "\n" ) if ( nfa_mod > 0.3 ) { cat( "👉 模块度 > 0.3,网络社区结构显著\n" ) } else { cat( "👉 模块度 ≤ 0.3,网络社区结构较弱\n" ) } cat( "=====================================\n" ) V( g) $ community <- nfa_best cat( "\n各社区基因数量:\n" ) print( table( nfa_best) ) out_dir <- "data/NFA_Community_Proteins" if ( ! dir.exists( out_dir) ) { dir.create( out_dir) } gene_name <- names ( V( g) ) cluster_id <- membership( nfa_comm) gene_cluster_df <- data.frame( Gene = gene_name, Cluster = cluster_id, row.names = NULL ) %>% arrange( Cluster) write.table( gene_cluster_df, file = "data/NFA_Community_Proteins/a_Cluster_Info.txt" , sep = "\t" , quote = FALSE , row.names = FALSE , col.names = FALSE ) cat( "\n✅ 已输出:a_Cluster_Info.txt (第1列基因名,第2列集群编号)\n" ) comm_ids <- unique( nfa_best) for ( cid in comm_ids) { node_in_comm <- names ( nfa_best[ nfa_best == cid] ) file_path <- file.path( out_dir, paste0( "community_" , cid, ".txt" ) ) writeLines( text = node_in_comm, con = file_path) } cat( "\n✅ 已新建文件夹【NFA_Community_Proteins】,所有聚类文件已存入该文件夹\n" ) global_dens <- edge_density( g) member_all <- membership( nfa_comm) keep_comm <- c ( ) for ( comm_id in unique( member_all) ) { nodes_idx <- which( member_all == comm_id) if ( length ( nodes_idx) > 5 ) { subg <- induced_subgraph( g, nodes_idx) comm_dens <- edge_density( subg) if ( comm_dens > global_dens) { keep_comm <- c ( keep_comm, comm_id) } } } plot_nodes <- which( member_all %in% keep_comm) g_plot <- induced_subgraph( g, plot_nodes) V( g_plot) $ community <- member_all[ plot_nodes] deg <- degree( g_plot) V( g_plot) $ size <- sqrt ( deg) * 1.2 n_comm <- length ( unique( V( g_plot) $ community) ) pal <- rainbow( n_comm) V( g_plot) $ color <- pal[ as.factor( V( g_plot) $ community) ] E( g_plot) $ color <- "gray30" E( g_plot) $ width <- 1 node_names <- names ( V( g_plot) ) V( g_plot) $ short_label <- substr( node_names, nchar( node_names) - 3 , nchar( node_names) ) lay <- layout_with_fr( g_plot) scale_factor <- 3.5 lay <- lay * scale_factor plot( g_plot, main = "基因互作网络 NFA 社区聚类图" , layout = lay, vertex.label = V( g_plot) $ short_label, vertex.label.cex = 0.28 , vertex.label.dist = 1.1 , vertex.label.color = "black" , edge.color = E( g_plot) $ color, edge.width = E( g_plot) $ width, margin = c ( 0.4 , 0.4 , 0.4 , 0.4 ) ) png( "png/GeneNetwork_NewmanFast_protein.png" , width = 1800 , height = 1800 , res = 150 ) plot( g_plot, main = "基因互作网络 NFA 社区聚类图" , layout = lay, vertex.label = V( g_plot) $ short_label, vertex.label.cex = 0.28 , vertex.label.dist = 1.1 , vertex.label.color = "black" , edge.color = E( g_plot) $ color, edge.width = E( g_plot) $ width ) dev.off( ) cat( "\n✅ 网络图已保存到当前文件夹:GeneNetwork_NewmanFast_protein.png\n" )
statistic_NFA_.R 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 library( igraph) library( dplyr) library( readr) set.seed( 123 ) edge_file <- "data/511145.protein.physical.links.v12.0.min900.txt" cluster_file <- "data/NFA_Community_Proteins/a_Cluster_Info.txt" edges <- read_delim( edge_file, delim = " " , col_names = FALSE , show_col_types = FALSE ) %>% select( 1 , 2 ) colnames( edges) <- c ( "protein1" , "protein2" ) clusters <- read_delim( cluster_file, delim = "\t" , col_names = FALSE , show_col_types = FALSE ) colnames( clusters) <- c ( "protein" , "module" ) g <- graph_from_data_frame( edges, directed = FALSE ) g <- simplify( g, remove.multiple = TRUE , remove.loops = TRUE ) V( g) $ module <- clusters$ module[ match( V( g) $ name, clusters$ protein) ] g <- induced_subgraph( g, ! is.na ( V( g) $ module) ) cat( "=== 网络基础信息 ===\n" ) cat( "总节点数:" , vcount( g) , "\n" ) cat( "总边数:" , ecount( g) , "\n" ) nfa_module_num <- length ( unique( V( g) $ module) ) cat( "总模块数(NFA划分):" , nfa_module_num, "\n\n" ) cat( "=== NFA全局模块显著性分析 ===\n" ) real_module <- make_clusters( g, V( g) $ module) real_Q <- modularity( real_module) cat( "真实网络 NFA 模块化度 Q =" , round ( real_Q, 4 ) , "\n" ) n_perm <- 1000 random_Q <- numeric( n_perm) for ( i in 1 : n_perm) { g_random <- rewire( g, keeping_degseq( niter = vcount( g) * 5 ) ) nfa_hc <- cluster_fast_greedy( g_random) memb_vec <- cut_at( nfa_hc, no = nfa_module_num) rand_clu <- make_clusters( g_random, memb_vec) random_Q[ i] <- modularity( rand_clu) if ( i %% 200 == 0 ) cat( "已完成" , i, "次随机置换检验\n" ) } p_raw <- mean( random_Q >= real_Q) p_value <- ifelse( p_raw == 0 , 1 / ( n_perm + 1 ) , p_raw) cat( "全局显著性 P值 =" , ifelse( p_raw == 0 , paste0( round ( p_value, 4 ) , " (P < 0.001)" ) , round ( p_value, 4 ) ) , "\n" ) cat( "随机网络平均 Q =" , round ( mean( random_Q) , 4 ) , "\n" ) cat( "随机网络 Q 标准差 =" , round ( sd( random_Q) , 4 ) , "\n" ) cat( "Z-score =" , round ( ( real_Q - mean( random_Q) ) / sd( random_Q) , 4 ) , "\n" ) cat( "结果判定:" , ifelse( p_value < 0.05 , "✅ NFA模块结构显著(非随机)" , "❌ NFA模块结构无统计学显著性" ) , "\n\n" ) write.csv( data.frame( random_Q) , "data/NFA_Community_Proteins/random_Q.csv" , row.names = FALSE ) module_stats <- lapply( unique( V( g) $ module) , function ( m) { module_nodes <- V( g) $ name[ V( g) $ module == m] subg <- induced_subgraph( g, module_nodes) intra_edges <- ecount( subg) total_edges_all <- ecount( g) data.frame( 模块编号 = m, 节点数 = vcount( subg) , 边数 = ecount( subg) , 模块密度 = round ( edge_density( subg) , 4 ) , 平均度 = round ( mean( degree( subg) ) , 2 ) , 连通分量数 = components( subg) $ no, 内部边比例 = round ( intra_edges / total_edges_all, 4 ) ) } ) %>% bind_rows( ) %>% arrange( desc( 模块密度) ) net_density <- edge_density( g) key_modules <- module_stats %>% filter( 节点数 >= 5 , 模块密度 > net_density, 连通分量数 == 1 ) cat( "=== NFA筛选关键模块列表 ===\n" ) print( key_modules) write_delim( arrange( module_stats, 模块编号) , "data/NFA_Community_Proteins/aNFA_蛋白质模块全统计结果.txt" , delim = "\t" ) write_delim( arrange( key_modules, 模块编号) , "data/NFA_Community_Proteins/aNFA_关键蛋白质模块筛选结果.txt" , delim = "\t" ) cat( "\n✅ NFA方法专属分析完成!统计结果已保存至 NFA_Community_Proteins 目录" ) plot_df <- data.frame( Q_value = random_Q) p <- ggplot( plot_df, aes( x = Q_value) ) + geom_histogram( aes( y = ..density..) , bins = 30 , fill = "#87CEEB" , alpha = 0.6 , color = "white" ) + geom_density( color = "#1f77b4" , linewidth = 1 ) + geom_vline( xintercept = real_Q, color = "#e74c3c" , linewidth = 1.2 , linetype = "solid" ) + geom_vline( xintercept = mean( random_Q) , color = "#2c3e50" , linewidth = 1 , linetype = "dashed" ) + annotate( "text" , x = real_Q, y = max ( density( random_Q) $ y) * 0.8 , label = paste0( "Observed Q = " , round ( real_Q, 4 ) ) , color = "#e74c3c" , hjust = - 0.05 , size = 3.5 ) + annotate( "text" , x = mean( random_Q) , y = max ( density( random_Q) $ y) * 0.6 , label = paste0( "Mean random Q = " , round ( mean( random_Q) , 4 ) ) , color = "#2c3e50" , hjust = 1.05 , size = 3.5 ) + annotate( "text" , x = max ( random_Q) , y = max ( density( random_Q) $ y) * 0.4 , label = paste0( "P = " , round ( p_value, 4 ) , "\nZ = " , round ( ( real_Q - mean( random_Q) ) / sd( random_Q) , 4 ) ) , size = 3.5 , hjust = 1 ) + labs( x = "Modularity Q" , y = "Density" , title = "NFA Community Permutation Test (1000 Randomizations)" ) + theme_bw( ) + theme( plot.title = element_text( hjust = 0.5 , size = 12 ) , axis.title = element_text( size = 11 ) , panel.grid = element_blank( ) ) print( p) ggsave( "png/NFA_Permutation_Test_Q.png" , plot = p, width = 6 , height = 4.5 , dpi = 300 )
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 import pandas as pddf=pd.read_csv('../data/DNE_cengci_Community_Proteins/aDNE_关键蛋白质模块筛选结果.txt' ,sep='\t' ) df_filter=df['模块编号' ] df_filter.head() afewmodules_name=[] for i in range (len (df_filter)): module=df_filter[i] afewmodules_name.append("community_" + str (df_filter.iloc[i])+".txt" ) afewmodules_name[0 ] for modele in afewmodules_name: with open ('../data/DNE_cengci_Community_Proteins/' +modele) as f: lines=f.readlines() lines=[line[7 :12 ] for line in lines] with open ('../data/filter_DNE/' +modele,'w' ) as f1: for line in lines: f1.write(line+'\n' )
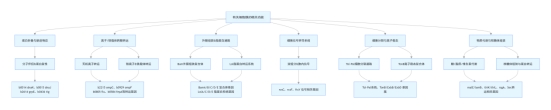
readme.md 蛋白质模块发现实验(Submit) 本目录为“蛋白质模块发现”实验的提交结果目录,包含代码、数据和可视化输出,主要用于复现实验流程和结果。
目录结构